


アンジェルマン症候群(Angelman syndrome、AS: OMIM 105830)
疾患概要
ASは、重度発達遅滞、てんかん、失調性運動障害、容易に引き起こされる笑い、下顎突出を含む特徴的な顔貌などを特徴とする症候群です(Williams et al. Genet Med. 2010)。原因遺伝子UBE3Aはインプリンティング領域である15q11-q13に存在し、神経細胞において母由来アレルのみが発現するインプリンティング遺伝子です。多くは孤発例ですが、稀に家族例が存在し、その場合は母親が保因者となり得ます。同じ15q11-q13に存在する父性発現遺伝子が原因のプラダー・ウイリー症候群(Prader-Willi syndrome, PWS)と関連が強い疾患ですが、臨床症状は大きく異なります。
発症頻度
ASの頻度は約1万5千出生に1人であり、男女差はありません。比較的頻度の高い遺伝性疾患のひとつです(Williams et al. Genet Med. 2010)。
臨床像
発達の遅れは乳児期にははっきりせず、4か月健診で気づかれることは稀です。発達の遅れは10か月頃までに明らかになります。歩行開始の平均年齢は5歳と報告されています。発達の遅れは重度であり、有意語の獲得はほとんどみられません。言語理解より言語表出の遅れが目立ち、発語はなくても、年齢とともに理解は進みます。てんかんは80%程に合併し、乳児期に熱性けいれんとして発症する場合が多く、その後無熱性発作が出現します。てんかん発作は全般発作(全身けいれんやミオクロニー発作など)が多いですが、意識消失発作や焦点発作もみられます。非けいれん性てんかん重積はASの特徴のひとつとして言われ、頻度は高くないですが、重要なてんかん発作です。行動特徴として、容易に引き起こされる笑いの他に、多動や好奇心、水やビニールなどのキラキラしたものに対する興味が認められます。乳児期から睡眠障害の合併がみられ、夜間中途覚醒や遅い入眠と早期覚醒を認めます。特徴的な顔貌として、尖った下顎と大きな口があります。低色素症の合併があると、新生児期から色白が目立ち、頭髪が薄い茶色を示します。低色素症は年齢とともに目立たなくなります。成人になると活動性が低下して、歩行能力を失うことがあり、肥満の合併も多くみられるため、注意が必要です。
遺伝学的原因
ASは15番染色体q11-q13に位置するUBE3Aの機能喪失により発症します(図1)。UBE3A蛋白はユビキチン蛋白リガーゼであり、標的蛋白をユビキチン化し、分解する働きがあります。UBE3Aの働きが失われると、標的蛋白が蓄積することで機能障害を引き起こすと考えられています。ここで、UBE3Aは神経細胞において母由来アレルのみが発現しているインプリンティング遺伝子です。そのため、母由来UBE3Aの機能喪失が疾患の原因となります。神経細胞以外ではUBE3Aはインプリンティングを受けていず、両親由来の遺伝子が発現しています。そのため、ASは中枢神経だけの症状を示します。中枢神経における標的蛋白として複数の蛋白が同定されています。その中のひとつであるArcは興奮性神経伝達物質であるグルタミン酸の受容体のひとつであるAMPA型受容体を調節している蛋白です。UBE3Aの機能喪失はArc蛋白の増加を介してAMPA型受容体を減少させ、グルタミン酸シナプスの機能障害を引き起こし、その結果、経験依存性シナプス可塑性が障害されると考えられ、知的障害の原因のひとつと考えられています(Greer et al. Cell. 2010)。
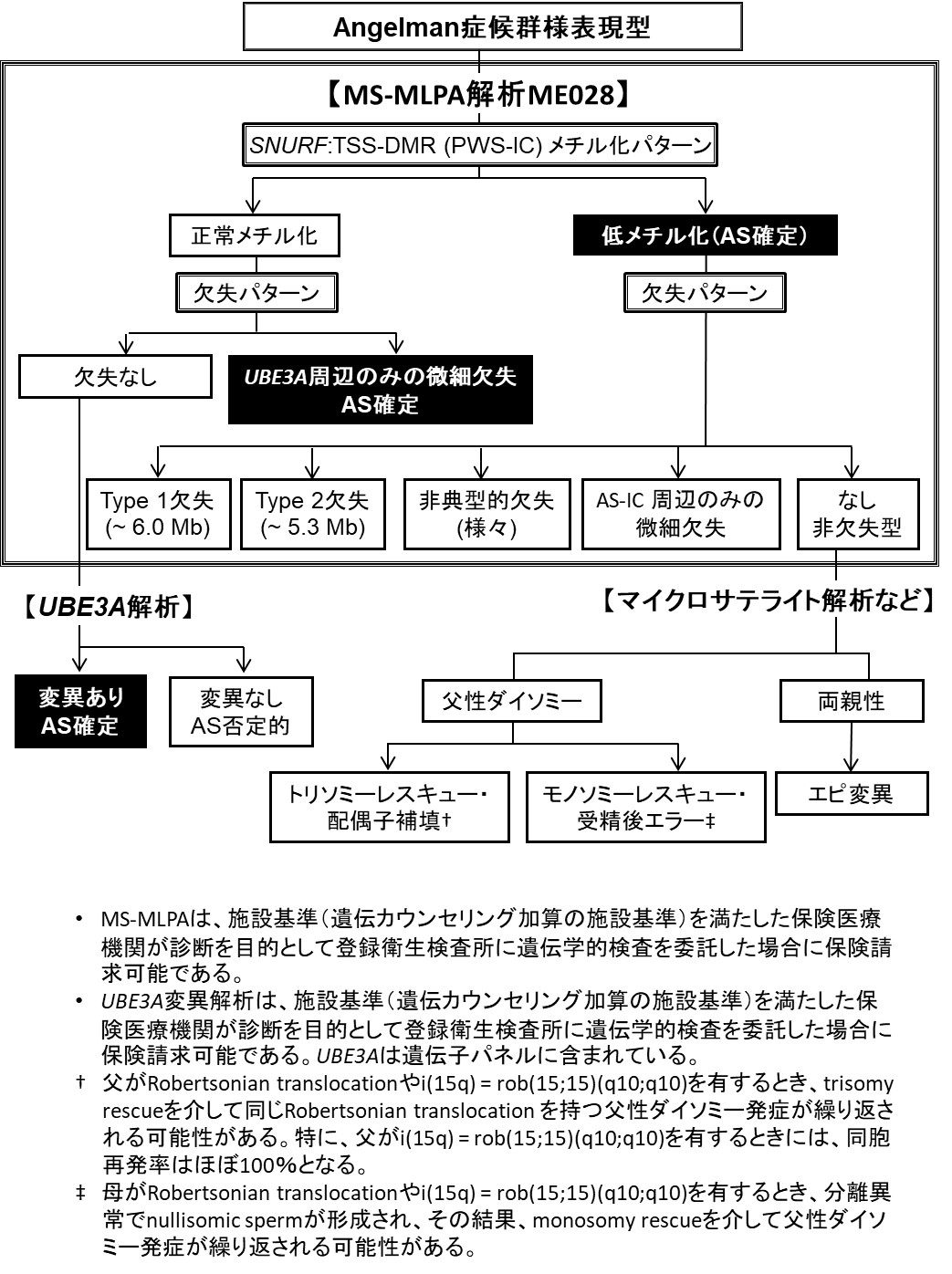
UBE3Aの機能喪失が起こるメカニズムは以下の4つが知られています(図2)。
- ①15q11-q13の母性欠失(70%)
②15番染色体の父性片親性ダイソミー(uniparental disomy: UPD)(5%)
③刷り込み機構の異常である刷り込み変異(5%)
④UBE3A遺伝子の変異(10%)
残り10%では明らかな原因が同定されません(クラスV)。
前述したようにUBE3Aはインプリンティング遺伝子です。インプリンティングもしくはゲノムインプリンティングとは親由来により遺伝子の発現が異なる遺伝現象です。インプリンティングはDNA配列に依存しない、エピジェネティックな現象です。ほとんどの遺伝子はインプリンティングを受けていず、両親由来どちら遺伝子も同様に発現しています。インプリンティング遺伝子はまとまって、染色体の一部の領域に存在することが知られており、前述したようにUBE3Aが存在する15q11-q13はよく知られたインプリンティング領域です。15q11-q13にはUBE3A以外に複数のインプリンティング遺伝子が存在します(図1)。UBE3Aは母性発現遺伝子ですが、父性発現遺伝子も複数存在し、これらの機能障害がPWSの原因となることが知られています。以下にそれぞれの遺伝学的原因について解説いたします。
- 15q11-q13の母性欠失(70%)
ASの大部分(70%)を占めるのが、約4Mbの欠失です。欠失の切断点にリピート構造存在するために、欠失範囲はほとんどの例で共通しPWSに認める欠失の範囲と同じです(図2)。ただ、ASでは欠失は例外なく母由来染色体であり、PWSでは父由来染色体です。UBE3Aは神経細胞で母由来のみ発現しているため、母由来欠失では神経細胞におけるUBE3Aの発現がなくなります。PWSでは父由来染色体での欠失なので、母由来のUBE3Aの発現は保たれ、その代わり父性発現遺伝子(SNRPN、SNPRD116、MAGEL2など)の発現が消失し、PWSを発症します。欠失の中には両親性発現遺伝子が存在し、これらの発現は半分になります。両親性発現遺伝子の一つであるGABRB3の欠失はてんかんを含めたASの重症度に影響すると考えられています。また、両親性発現遺伝子P (OCA2)の欠失は低色素症との関連が知られています。そのため、ASでは欠失型が最も重症の表現型を示します(Fujimoto et al. HGG Adv. 2024)。
- 15番染色体の父性片親性ダイソミー(uniparental disomy: UPD)(5%)
2本の15番染色体が両方父親由来となる現象を父性片親性ダイソミー(UPD)と言います。15番染色体父性UPDでは母由来染色体が存在しないため、母性発現を示すUBE3Aの発現は失われ、ASが発症します。15番染色体母性UPDはPWSの原因となります。PWSでは母性UPDは25%を占めますが、ASでは5%にとどまります。PWSでは母親の減数分裂における15番染色体の不分離が大元の原因であり、15番染色体のトリソミー細胞(胎生致死)から発生早期に父由来15番染色体が失われるメカニズム(トリソミーレスキュー)が大部分を占めます。これに対して、父親の精子形成過程では不分離の確率が卵子形成過程よりは格段に低いため、トリソミーレスキューの頻度が低いと考えられています。代わりに、母親の減数分裂における不分離の結果発生しうる、15番染色体のモノソミー(胎生致死)が発生早期に倍加するメカニズム(モノソミーレスキュー)もしくは、正常受精後の発生早期での体細胞分裂における不分離(モザイク15番染色体トリソミー)からのトリソミーレスキューの可能性が指摘されています(Fujimoto et al. J Hum Genet. 2023)。これらのメカニズムはPWSにおけるUPD発症メカニズムと比較して複雑なため、ASでのUPDの頻度がPWSと比較して低いと考えられています。
刷り込み変異は15番染色体が両親由来であるにも関わらず、15q11-q13のインプリンティングが異常となる状態です。ASの刷り込み変異では、母由来15番染色体の15q11-q13のインプリンティングの状態(DNAメチル化や遺伝子発現)が父由来のパターンになっています。そのため、母由来UBE3Aの発現が失われ、ASが発症します。刷り込み変異の10%には刷り込み中心(imprinting center: IC)と呼ばれる領域(AS-IC)の微細欠失が母由来染色体に同定されます。AS-ICは卵子形成過程で母由来インプリンティングを形成するために必須な領域と考えられており、AS-ICが欠失すると母性インプリンティングの形成ができず、父由来パターンになると考えられています。AS-ICは父性インプリンティング形成には必要がなく、AS-ICがなくても父性インプリンティングが形成できることが示されています。刷り込み変異の90%ではAS-ICの欠失は同定されません。これらの患者はエピ変異と呼ばれます。エピ変異は何らかのエピジェネティックな原因でインプリンティングの形成ができないと考えられていますが、未だそのメカニズムは不明です。PWSにも刷り込み変異が存在します。PWS-ICの場所はAS-ICと非常に近接していますが、異なっています。PWSでも刷り込み変異の中で、PWS-IC欠失例は10%ほどで、90%はエピ変異です。ASでもPWSでもエピ変異例には遺伝性はありません。しかし、AS-IC、PWS-ICの微細欠失例では、AS-ICは母性刷り込み、PWS-ICは父性刷り込みの形成にのみ影響があるので、AS-IC微細欠失が父由来、PWS-IC欠失が母由来の場合はインプリンティングへの影響はなく保因者となります。そのため、家族例が存在し、遺伝性が考慮されます(Nicholls et al. Trends Genet. 1998)。
- UBE3A遺伝子の変異(10%)
母由来UBE3Aに機能喪失型バリアントが存在すると、ASの原因となります。同じバリアントを持つ母親は、そのバリアントが母型祖父由来の場合は、病因とならず、保因者となります。従って、UBE3A遺伝子変異例では遺伝性を考慮することが必要になります。UBE3Aの病因バリアントは様々な種類があるため、表現型の幅はやや広くなります。一般的にはUBE3A変異例は欠失例よりは表現型が軽い傾向があります(Fujimoto et al. J Hum Genet. 2023)。
遺伝学的診断
国際的にASの診断の第一選択は15q11-q13 のDNAメチル化テストです。DNAメチル化テストでは15q11-q13の親由来パターンがわかるため、欠失、UPD、刷り込み変異を同定することができます。DNAメチル化テストが陽性の場合は、欠失、UPD、刷り込み変異の区別が必要となります。欠失とUPDは原則として遺伝性はありませんが、刷り込み変異では10%が家族例です。欠失はFISH法で同定が可能であり、マイクロアレイ染色体検査やMLPA法でも同定が可能です。後者2つでは欠失の範囲を同定できる利点があります。DNAメチル化テストとMLPA法を組み合わせたMS-MLPA法を用いると、DNAメチル化テストとMLPA法が同時に実施できるため、欠失、UPDもしくは刷り込み変異の診断が一回でできます。国立成育医療研究センター衛生研究所において保険診療で実施が可能になっており、刷り込み変異の微細欠失の同定も可能です。しかし、UPDと刷り込み変異を完全に区別するためには、両親の検体を用いた多型解析が必要になります(非保険)。DNAメチル化テスト、FISH法、マイクロアレイ染色体検査とMLPA法は保険適応となっています。UBE3A変異は遺伝子のシークエンスで解析し、保険診療での実施が可能です(かずさDNA研究所)。
管理・治療法
ASの本質的な治療法はありません。しかし、UBE3Aの遺伝子発現を回復させるアンチセンス核酸療法や遺伝子治療などが開発されており、期待されています(Meng et al. Nature. 2015)。現時点では、対症療法が重要です。てんかんに対する抗てんかん薬、睡眠障害に対するメラトニン製剤の使用が比較的多くの患者で行われています。専門医などと連携した治療が望まれます。また、ASは乳児期から生涯にわたり支援が必要な疾患です。そして、小児慢性特定疾病、指定難病に指定されています。必要な手続きを行い、多職種と連携することで、QOLを支える支援が望まれます。
予後
予後は良好とされていますが、成人期以降は活動性の低下や肥満などへの対応が必要になることがあります。
小児、指定難病ページ、各大学、関連学会へのリンク
患者会
図の説明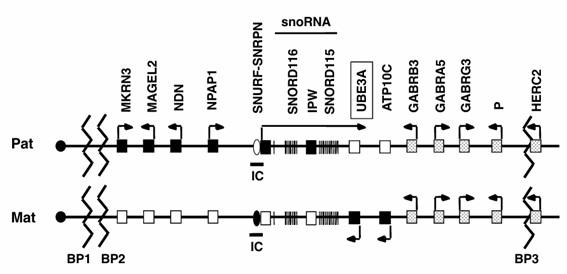
図1. 15q11-q13の遺伝子地図
Pat:父由来染色体、Mat:母由来染色体。AS原因遺伝子UBE3Aを枠で囲んだ。■は活性遺伝子で→は転写の方向を示す。□は不活性遺伝子、□の中のドットは両親性発現遺伝子を示す。ジグザク線は欠失の切断点(BP)を示す。IC:刷り込み中心。
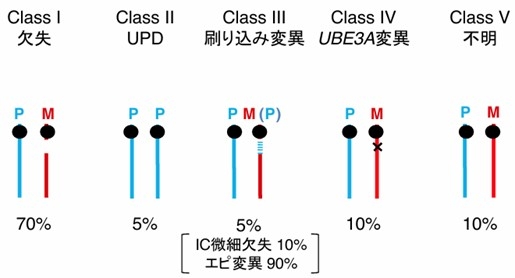
図2. ASの遺伝学的分類