


- トップ
- > 研究者・企業の方へ
- > 研究所について
- > 各研究部門の紹介
- > 分子内分泌研究部
- > プラダー・ウイリ症候群(Prader-Willi syndrome、PWS: OMIM 176270)
プラダー・ウイリ症候群(Prader-Willi syndrome、PWS: OMIM 176270)
疾患概要
PWSは、1956年内分泌科医のプラダーと神経科医のウイリーが報告した疾患で、最初にみつかったインプリンティング疾患として有名である。遺伝子は、通常、親由来にかかわらず同様に働くが、例外的に、父親由来のときのみ働く遺伝子(父性発現遺伝子)や母親由来のときのみ働く遺伝子(母性発現遺伝子)が存在する。ここで、インプリントとは、遺伝子の発現を抑制するマーキングのことであり、ゲノム配列の変化ではなく、CpG配列 (CpG islands)のメチル化などの可変的な修飾によるものであり、このためにエピジェネティクス(エピ:後成的)という用語が用いられる。ここでは、SNRPN遺伝子上流のメチル化可変領域(DMR: differentially methylated region)が、インプリンティングセンターとして作用し、このDMRは父由来のとき非メチル化状態、母由来のときメチル化包帯で存在する。このメチル化状態がインプリンティングの維持に必須であるため、このDMRはインプリンティングセンターとも呼ばれる。
発症頻度
出生児の約15,000人に1人とされているが、実際にはもっと少ないと思われる。
臨床像
プラダー・ウイリ症候群の症状は、内分泌学的異常(肥満、低身長、性腺機能障害、糖尿病など)、神経学的異常(筋緊張低下、特徴的な性格障害、異常行動)、体組成異常(筋肉量の減少や体脂肪の増加)を主体として、多岐にわたり、かつ年齢に応じて変化する。新生児期は、筋緊張低下、色素低下、外性器低形成を3大特徴とする。筋緊張低下が顕著で哺乳障害のため経管栄養となることが多い。色素低下の顕著な患者では頭髪は金髪様となり白皮症と誤診される場合もある(この色素低下は、欠失タイプ遺伝子特徴的であり、これは、両親性発現をする色素に関連する遺伝子が欠失することによる)。外性器低形成として、男児では停留精巣やミクロペニスが90%以上に認められるが、女児では陰唇あるいは陰核の低形成は見逃されやすい。3~4歳頃から過食傾向が始まり、幼児期には肥満、低身長が目立ってくる。学童期には、学業成績が低下し、性格的にはやや頑固となってくる。思春期頃には、二次性徴発来不全、肥満、低身長、頑固な性格からパニック障害を示す人がいる。思春期以降、肥満、糖尿病、性格障害・行動異常などが問題となる。とりわけ、性格障害・異常行動は、患者本人あるいは家族が一番悩まされる事象である。性格は、年齢を経るに従い、可愛いから、しつこい、頑固、パニック、暴力へとエスカレートすることがあり、行動異常では、万引き、嘘を言うなどの反社会的行動が目立ち、社会の中で上手くやっていけない場合がある。その原因は、不明であるが、多くの患者が酷似した性格傾向を示すことから、遺伝的背景の関与が示唆される。 このように症状は多彩であるが、その病因は間脳の異常に集約される。間脳には、種々の中枢が存在し、食欲中枢(過食、肥満の原因)、呼吸中枢(中枢性無呼吸や昼間の過度の睡眠の原因)、体温中枢(冬場の低体温、夏場の高体温)、情緒の中枢(性格障害との関連)、性の中枢(二次性徴発来不全の一因)、など間脳の異常に起因した多彩な症状の説明が可能である。
遺伝学的原因
PWSは、染色体15q11-13領域の父性発現遺伝子が作用しなくなることで発症する。特にSNORD116の発現消失が、最も重要な役割を果たしている。約75%が欠失(上記インプリンティング領域の欠失で、2つの欠失がほとんどの症例で同定されている)、約20%が母性片親性ダイソミー(1対の第15染色体が共に母親に由来する状態)に起因し、残る少数例は、エピ変異(上記の父由来DMRがメチル化された状態)、SNORD116周辺のみの欠失、インプリンティングセンターとSNORD116の連続性を破断する転座や逆位、ホスト遺伝子であるSNEPNの機能低下変異に起因する。片親性ダイソミーは、trisomy rescue、gamete complementation、monosomy rescue、postfertilization errorにより発症し、高齢出産は第一減数分裂の不分離に起因するtrisomy rescue発症リスクとなる。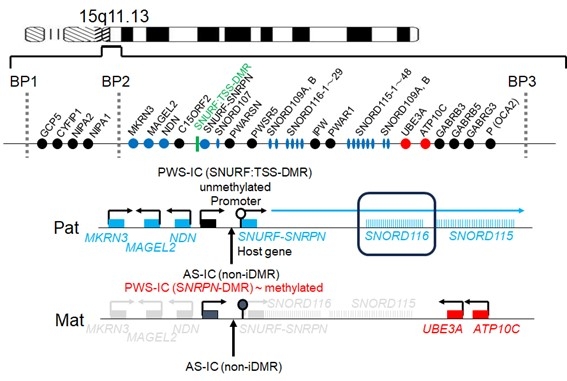
遺伝診断法および遺伝学的診断機基準
保険適用となっている遺伝学的検査には、メチル化試験、MS-MLPA、アレイCGH、FISH検査が含まれる。アレイCGHとFISH検査は欠失型しか診断できないが、メチル化試験とMS-MLPAは欠失型、母性片親性ダイソミー、エピ変異の全てを診断できる。さらにMS-MLPAは、SNORD116を含むがインプリンティングセンターであるSNURF:TSS-DMRを含まない微細欠失、SNURF:TSS-DMR周辺のみの微細欠失、ならびに一定頻度以上の母性片親性ダイソミーモザイクも検出できる(最新の「プラダー・ウイリ症候群コンセンサスガイドライン」を参照:日本小児内分泌学会ホームページ(http://jspe.umin.jp/medical/gui.html)。したがって、MS-MLPAが第1選択の検査として推奨される。この解析は、施設基準を満たした保険医療機関がPWSの診断を目的として登録衛生検査所(現時点では国立成育医療研究センターのみ)に遺伝学的検査を委託した場合に保険請求可能である。正常核型のPWS患者を対象とする診断フローチャートを示す。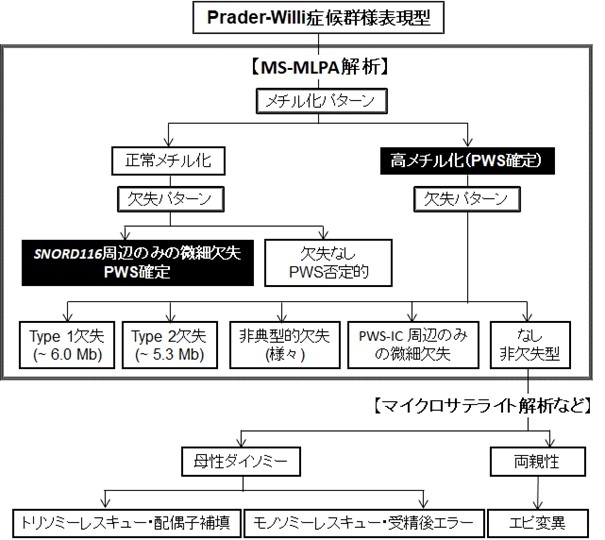
MS-MLPAは、1回の検査で、PWSの診断に加えて、欠失型と非欠失型(大多数は母性ダイソミー、少数がメチル化異常)の鑑別のみならず、モザイクもある程度まで解析可能であり、さらに、欠失型では、コモン欠失 (Type 1とType 2) のみならず、非典型的欠失、SNORD116周辺のみの欠失、PWS-ICのみの欠失を同定できる(上図の四角に囲まれた部分)。ここで、コモン欠失は、前述の疾患概要のところに記載した図に示す250 - 400 kbのlow-copy repeatsに介在される欠失を指し、BP1-BP3に介在される約6.0 MbのType 1欠失が約36%を、BP2-BP3に介在される約5.3 MbのType 2欠失が約56%占める。非欠失型は、母性ダイソミーとエピ変異からなる。そして、母性ダイソミーは、さらにいくつかに細分され、このうち、卵形成第一減数分裂の不分離に起因するトリソミーレスキュー・配偶子補填タイプは、高齢妊婦からの出産児に多い。また、エピ変異は、ごく一部が一卵性双胎や卵母細胞・受精卵のメチル化を維持する遺伝子の変異に起因するmultilocus imprinting disturbanceとして発症することが判明しているが、大多数の症例では原因不明であり、受精後の有糸分裂時のエラーであると考えられている 。
以下、留意点を記載する。
- MS-MLPA解析により、Prader-Willi症候群の診断は、SNORD116より近位部のホスト遺伝子内に切断点を持つ転座 (および逆位) とホスト遺伝子の変異(SNRPNバリアントが報告されている)以外、可能である。なお、ダイソミーとエピ変異の鑑別には、マイクロサテライト解析やSNPアレイ解析などが必要である。
- 非典型的欠失には様々なタイプがあるが、特に重要なものは、コモン欠失よりも大きな欠失である。ここで、BP1より近位のMS-MLPAプローブはなく、BP3より遠位のプローブが1個のみ(APBA2)であり、このプローブで欠失が示され、非典型な症状(重度精神運動発達遅滞やてんかんなど)が見られるときには、より広範な欠失が示唆される。正確な欠失範囲の同定にはアレイCGHが推奨される。
- 染色体検査は以下の場合に考慮される。
- MS-MLPAでメチル化異常も欠失もなく、しかし、典型的なPWS症状が認められるとき: SNORD116より近位部のホスト遺伝子内に切断点を持つ転座 (および理論的には逆位も相当する)が報告されている。これは、SNORDがSNURF-SNRPNの発現に伴って転写されることから、両者の連続性の破壊がSNORD116発現の低下・消失を招くためとされている。
- トリソミーレスキュー・配偶子補填タイプの母性ダイソミーのとき:母がRobertsonian translocationやi(15q) = rob(15;15)(q10;q10)を有するとき、trisomy rescueを介して同じRobertsonian translocation を持つ母性ヘテロダイソミー発症が繰り返される可能性がある。特に、母がi(15q) = rob(15;15)(q10;q10)を有するときには、同胞再発率はほぼ100%となる。
- モノソミーレスキュータイプの母性ダイソミーのとき:父がRobertsonian translocationやi(15q) = rob(15;15)(q10;q10)を有するとき、分離異常でnullisomic spermが形成され、その結果、monosomy rescueを介して母性アイソダイソミー発症が繰り返される可能性がある。
- コモン欠失より大きな欠失が存在するとき:この場合、インプリンティング領域外に切断点を有する転座保因者である父に由来する不均衡型転座の可能性がある。
- シークエンス検査は以下の場合に考慮される。
- MS-MLPAや染色体検査で異常がなく、しかし、典型的なPWS症状が認められるとき: SNORD116より近位部のホスト遺伝子内変異 (SNRPN変異) が報告されている。
管理・治療法
本症は、病因が染色体異常のため根本的治療法はない。かつ、症状が多岐に及ぶため多種の分野の専門家(小児科医、内分泌科医、遺伝科医、精神科医、臨床心理士、栄養士、教職員、理学療法士など)の協力による包括医療の重要性が強調されている。代表的な治療法は、以下の通りである。 1. 食事療法:本症では終生誰かが管理する必要のある一番大切で基本的治療法である。食事制限は2歳頃までは健常児と同じ、3~4歳頃から身長1cmあたり10 Kcalを目安に摂取カロリーの制限を行う必要がある。成人PWS患者の最終身長が約150 cmあたりのため、成人での摂取カロリーは1500 kcalが目安となる。大切なことは、彼らの手の届く所に安易に食べ物を放置しない事である。また、彼らは、摂取カロリーが多くなくても肥満になり易い傾向があることを周囲が良く認知し(体脂肪の動員が下手で、基礎代謝率も低い)、彼らの肥満に対して偏見を持たないようすることが不可欠である。
- 運動療法:体重維持に予想以上に貢献する。彼らが、捻挫などで通常の運動が不可能なとき、驚くほど短期間に体重が増加することは良く経験される事実である。彼らは、元来筋緊張低下があり運動は不得意であるが、現在まで多くの患者が水泳を取り入れることで運動療法が比較的成功している。脂肪の多い彼らの体組成は、水泳には向いていると考えられる。運動を強要するのではなく、一緒に運動に付き合うことも大切である。
- 成長ホルモン療法:現在世界的に実践されている治療法であり、本治療法がPWS患者の自然歴を大きく改善させてきている。成長ホルモンによる身長促進、体組成改善、筋力向上などは、すでに周知のこととなっている。今や世界中の関心は、成長ホルモン療法が直接あるいは間接的に知能や性格に及ぼす可能性に注目してきているが、それらの客観的評価は未だ難しい。成長ホルモン治療に伴って危惧されてきている問題点は、糖質代謝、呼吸障害、側弯症の3点に集約される。糖質代謝では、本症への成長ホルモ認可以前から成長ホルモンが糖尿病誘発する可能性が危惧されたが、実際は成長ホルモン治療で筋肉量増加、活動性向上のためインスリン感受性が改善しむしろ血糖が低下するといったデータのみが報告されており、現在、基本的食事療法が良好維持されている条件下では糖尿病誘発可能性はないと考えられている。呼吸障害に関しては、成長ホルモンは間脳にある呼吸中枢には好影響(中枢の酸素や二酸化炭素濃度への感受性を改善する)が、閉塞性障害を悪化する恐れがあることが報告されている。すなわち、成長ホルモンは、水分貯留傾向やリンパ組織の増大を惹起し、上気道の狭窄症状を起こす可能性が危惧されている。その為、成長ホルモン開始初期、とりわけ使用開始4ヵ月位は呼吸症状に注意し、狭窄症状出現あるいは増悪時は、成長ホルモンの中止あるいは減量が推奨される。側弯症に関しては、成長ホルモン治療が発症・増悪因子となる可能性は多数の解析からほぼ否定されてきている。
- 性ホルモン補充療法:本症患者全員が持っている性腺機能不全に対する治療であるが、現実的には種々の理由で実施されていないのが実際である。特に男性では、男性ホルモン補充が、患者の攻撃性を増加する、行動異常を増悪することが、危惧され未だ世界中が躊躇している。しかし、この様な危惧を指示する報告はなく、学問的根拠はない。われわれの経験では、患者を十分選択し、信頼関係を確立した後での治療では、過激製の増悪はなく、むしろパニック障害の減少を認めている。本治療の目的は二次性徴発来不全に対するのみではなく、骨密度改善、さらには彼らの精神的効果が大きいと考えられる。男性ではエナルモン125ー250 mg/dose/月、 女性ではカウフマン療法が行われる。
- インスリン治療:プラダーウイリー症候群患者は、10歳頃から思春期にかけて糖尿病をしばしば発症する。このようなとき、経口糖尿病治療薬のみでコントロールすることは困難で、インスリンを必要とすることが多い。 6. 向精神薬:欧米では、積極的に精神科から向精神薬の投与が行われているが、まだまだ推奨可能な処方はない。現在SSRI(選択的セロトニン再吸収阻害剤)が比較的広く使用されており、一部の患者で効果を発揮している。また、パニック時にはリスパダールがしばしば有用である。
予後
患者の生命予後は不明である。死亡原因は、3歳までの乳幼児では、ウイルス感染時の突然死が多い。成人では、肥満、糖尿病に伴う合併症(蜂窩織炎、肺栓塞、腎不全、心不全、など)などで死亡する。そのため、肥満、糖尿病に罹患しなければ生命予後は比較的良いかもしれない。国内では、少なくとも2名が55歳以上で存命である。
小児、指定難病ページ、各大学、関連学会へのリンク
コンセンサスガイドライン
Prader-Willi syndrome | Genetics in Medicine
患者会