


- トップ
- > 研究者・企業の方へ
- > 研究所について
- > 各研究部門の紹介
- > 分子内分泌研究部
- > 偽性副甲状腺機能低下症(Pseudohypoparathyroidism、PHP: OMIM 103580, 603233)
偽性副甲状腺機能低下症(Pseudohypoparathyroidism、PHP: OMIM 103580, 603233)
疾患概要
PHPは、20q13.3に存在するGNAS領域の異常により、副甲状腺ホルモン(PTH)上昇、低Ca血症、高リン血症を呈する疾患です。PHPは、母由来アレルGNAS変異を有しオルブライト遺伝性骨異形成症(AHO)を示す1A型(PHP1A)と、GNASメチル化異常を有しAHOを示さない(一部示す)1B型(PHP1B)に分類され、発症原因により遺伝性が異なります。
また、父由来アレルにGNAS変異を有する場合はAHOのみを呈する偽性偽性副甲状腺機能低下症(PPHP)になります。ここでは、主にPHPについて記載します。
発症頻度
日本におけるPHPの頻度は約10万人に1人と報告されています(Takatani et al. J Epidemiol. 2023)。
臨床像と臨床診断基準
PHPは近位尿細管におけるPTHへの抵抗性により低カルシウム血症,高リン血症をきたします。PHPはさらに、低身長、円形顔貌、短指症、異所性骨化/骨腫を特徴とするAHOを高頻度に認めるPHP1Aと、AHOを伴わないPHP1Bに分けられます。しかし、AHOを一部伴うPHP1Bが報告され、表現型のみで両者を鑑別するのは困難と考えられます(Mantovani et al. Nat Rev Endocrinol. 2018)。また、Ellsworth-Howard試験はPHPの診断において推奨されておらず、PTH上昇が明らかであれば必要ありません。
PTH抵抗性
PTH抵抗性は、PTH受容体(Gタンパク質共役型受容体)の構成成分であるGsαの近位尿細管における機能不全によって、PTHのシグナル伝達が障害され生じます(図1)。後述するGNASインプリンティング障害によるPTH抵抗性は、出生後徐々に顕在化し、PTHの上昇、高リン血症、次いで低Ca血症がみられます。PTHとリン値の上昇が始まってから低Ca血症が始まるまで、通常4.5年の間隔があります(Gelfand et al. J. Pediatr. 2006)。CaとビタミンDの必要量が増加する小児期後期に低Ca症状が現れる傾向にあります(Linglart et al. Horm Res Paediatr. 2013)。
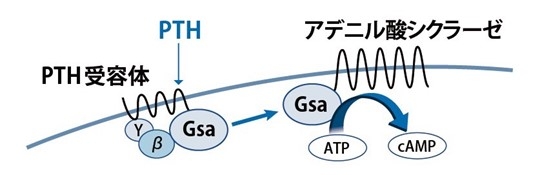
図1. PTHのシグナル伝達機構。PTHがPTH受容体に結合すると、三量体Gsタンパク質(α、β、ɤ)が活性化されます。活性化Gsαはアデニル酸シクラーゼに作用しcAMPを合成します。
AHO
AHOは、低身長、円形顔貌、短指症、異所性骨化/骨腫を特徴とする身体的特徴を指します(Mantovani et al. Nat Rev Endocrinol. 2018)。
低身長
PHP1Aの多くは、-2.5SD前後の成人身長を示します。子宮内発育遅延、GH放出ホルモン抵抗性、思春期以降の身長獲得の少なさなどが影響していると考えられています。一方で、PHP1Bの成人身長は一般集団とかわりません(Hanna et al. J Bone Miner Res. 2018)。
円形顔貌
罹患していない同胞との比較、あるいは全体的な肥満の程度と円形が目立つ場合あるいは肥満の程度と定義されます。PHP1Aの多くの症例、PHP1Bの30%程度に認められます(Urakawa et al. Eur J Endocrinol. 2023)。
短指症
PHP1Aでは70-80%、PHP1Bでは15-33%程度にみられます。第3、4、5中手骨と第1、4遠位指骨が最も影響を受け、幼少期には明らかでない場合もあります。
異所性骨化/骨腫
異所性骨化は間葉系幹細胞のGsα欠損の現れであり、Caおよびリンの血清レベルとは無関係です。PHP1Aの約半数で、出生時や生後早期からみられ、とくに短縮型変異症例で多く認められます。通常、PHP1Bではみられません(Urakawa et al. Eur J Endocrinol. 2023)。
乳幼児肥満
PHP1AおよびPHP1Bは、生後2年以内に肥満を起こすことがあります。視床下部におけるGsα依存性メラノコルチンシグナル伝達経路の障害による過食、安静時のエネルギー消費の減少、交感神経系活動の低下、脂肪分解の減少、成長ホルモン放出ホルモン抵抗性などいくつかのメカニズムが肥満の原因であると考えられています。年齢とともに、これらは徐々に改善し、成人期では肥満が目立たなくなります。したがって、特にAHOが目立たないPHP1Bにおいては、肥満が小児期唯一の症状である可能性があります(Urakawa et al. Eur J Endocrinol. 2023、Mantovani et al. Horm Res Paediatr. 2020)。
知的障害/発達障害
知的障害/発達障害は、PHP1Aの40-70%、PHP1Bの10%程度でみられ、重症度は非常に多様です。PHP1Aの平均IQは85.9で(Perez et al. Am J Med Genet A. 2018)、言語発達の方が運動発達より遅れる傾向にありますが、徐々にキャッチアップしていくと報告されています(Miyakawa et al. Endocr J. 2019)。PPHPではほとんど知的障害/発達障害がみられないことから、Gsαの脳でのインプリンティングが関与していると考えられています(Mantovani et al. Nat Rev Endocrinol. 2018)。
TSH・その他のホルモン抵抗性
TSH抵抗性は、甲状腺組織における部分的なGsαインプリンティングに起因し、PTH抵抗性ほど重度ではありません。PHP1AのほとんどはTSH抵抗性を呈し、PHP1Bでは、TSH値は正常値の上限か軽度の上昇を示します(Mantovani et al. Horm Res Paediatr. 2020)。日本においては、PHP1Bの38%がTSH抵抗性を有しており、その18%(全体の7%)が甲状腺ホルモンを投与されていました(Urakawa et al. Eur J Endocrinol. 2023)。そのほかに、PHP1Aではゴナドトロピン抵抗性、成長ホルモン放出ホルモン抵抗性(稀にPHP1B)を示すことがあります。
その他合併症
頭蓋内石灰化病変(異所性骨化とは異なります)、睡眠時無呼吸症候群、中耳炎、気管支喘息、歯科症状などがPHPと関連した合併症と考えられています(Mantovani et al. Nat Rev Endocrinol. 2018)。
遺伝学的原因
20q13.32に位置するGNAS遺伝子座は複雑なインプリンティング機構によって調節されたセンスおよびアンチセンス転写産物を生み出します。Gsαは、エクソン1-13によってコードされており、近位尿細管、甲状腺、生殖腺、下垂体などのいくつかの組織において、母由来アレルから優位に発現していますが、骨など他のほとんどの組織では両親性に発現しています。その他にGNAS遺伝子座からは、AS、神経内分泌分泌タンパク質55(NESP55)、XLαs、A/Bが発現し、それぞれAS-DMR、NESP-DMR、XL-DMR、A/B-DMRが調節しています。AS、XL、A/B-DMRは母由来アレルでメチル化されており、父由来アレルからのみ発現する父性発現遺伝子です。一方で、NESP-DMRは父由来アレルでメチル化されているため、母性発現遺伝子になります(図2)。NESP55、XLαs、およびA/Bのエクソン1はそれぞれ固有ですが、エクソン2-13はGsαコード領域と共通しています。Gsαをコードするエクソンのいずれかに変異がある場合、母由来アレルであればPHP1Aが発症し、父由来アレルであれば、 AHOのみを呈するPPHPが発症します。

図2. PHPの遺伝学的原因とGNAS遺伝子座DMRのメチル化状態。
一方、PHP1Bは、GNAS遺伝子座のメチル化異常によって発症し、さらに常染色体優性遺伝性PHP1B(AD-PHP1B)と孤発性PHP1B(Spor-PHP1B)に分類されます。発端者のみの頻度はAD-PHP1B:Spor-PHP1B=2:5でした(Urakawa et al. Eur J Endocrinol. 2023)。AD-PHP1Bの多くは、GNAS上流に位置するSTX16の母由来アレル微小欠失に起因し、A/B-DMRのみ低メチル化となります。STX16は両方の対立遺伝子から発現していますが、父由来アレルの欠失は表現型やメチル化状態に変化をもたらしません。NESP55からAS領域の欠失例やA/B上流へのレトロトランスポゾン挿入によるAD-PHP1Bも数例報告があり、それぞれDMRメチル化異常パターンは異なります(図2)。
Spor-PHP1Bは、20番染色体父性片親性ダイソミー(UPD(20)pat)とエピ変異に細分化されますが、DMRメチル化異常パターンは共通です。すなわち、A/B-DMR、XL-DMR、AS-DMRの低メチル化、NESP-DMRの高メチル化を認めます。Spor-PHP1BはGNAS領域の構造異常を認めず、次子あるいは次世代の再発リスクは、一般集団と同じで、無視しうる程度です。
近年、欧州のPHPコンソーシアムは、PHPを不活性化PTH/PTHrPシグナル伝達異常症(iPPSDs)と命名し、それぞれの基礎となる分子変化に応じて6つのサブグループに分類することを提案しました。iPPSD2は、PHP1AやPPHPを含むGsαの機能喪失変異によって定義されます。iPPSD3は、PHP1Bを含むGNAS遺伝子座の1つ以上のDMRのメチル化異常と定義されます(Thiele et al. Eur J Endocrinol. 2016)。
遺伝子診断法および遺伝学的診断基準
PHP1A、AD-PHP1B、Spor-PHP1B、PPHPとそれぞれ遺伝形式が異なるため、積極的な遺伝学的検査が望まれます。海外の報告では、AHOがあればGNAS変異解析、なければGNASメチル化解析が推奨されています(Mantovani et al. Nat Rev Endocrinol. 2018)。ただし、皮下骨腫/骨化を除き、PHP1BでもAHOがみられることがあるため、どちらを優先すべきかはケースバイケースになります。GNAS変異解析はかずさ遺伝子検査室(非保険)で行われています。GNASメチル化解析は、成育医療研究センター衛生検査センター(非保険)と当研究室(研究の一環)で行っております。
メチル化解析は、DMRのメチル化状態とコピー数変化を同時に解析することのできるメチル化特異的MLPA法(MS-MLPA)を用いて行います。A/B-DMRに低メチル化があれば、PHP1Bと診断できます。Spor-PHP1Bのメチル化異常パターンの場合は、トリオ検体でマイクロサテライトマーカー解析を行い、UPD(20)patかエピ変異かを同定します。
管理・治療法
低Ca血症に対しては、Ca補充や活性型ビタミンD製剤を投与します。成長ホルモン放出ホルモン抵抗性、TSH抵抗性、ゴナドトロピン抵抗性においても成長ホルモンや甲状腺ホルモン、性ホルモン補充療法を検討します。
予後
現在、PHP患者を根本的に治療する方法はなく、生涯にわたるフォローが必要になります。
小漫、指定難病ページ、各大学、関連学会へのリンク
・難病情報センター Pseudohypoparathyroidism, Type 1b (偽性副甲状腺機能低下症タイプIb
コンセンサスガイドライン
患者会
現時点ではありません。