


インプリンティング疾患
インプリンティング遺伝子とインプリンティング疾患
ゲノムインプリンティングという親由来を区別するためのマーキング(DNAメチル化やヒストン修飾)に従って親由来依存的に片親性に発現する遺伝子をインプリンティング遺伝子と呼び、父親由来の染色体上にある時のみ働く父性発現遺伝子(PEG: paternal expressed gene)と母親由来の染色体上にある時のみ働く母性発現遺伝子(MEG: maternally expressed gene)があります。通常の遺伝子は父由来、母由来ともに発現するので2コピーの発現となりますが、インプリンティング遺伝子はどちらかの親由来からのみ発現するので1コピーの発現となります。
インプリンティング遺伝子の過剰発現や発現消失はインプリンティング疾患を引き起こします。インプリンティング疾患は、通常の遺伝性疾患同様、遺伝子やゲノム構造異常でも発症しますが、インプリンティング遺伝子の発現パターンを調節しているマーキングの異常によっても発症し、このマーキングはエピ(エピ=後成的)ジェネティクスと表現されます。
インプリンティング領域とメチル化可変領域 (DMR)
インプリンティング遺伝子は、一群となって染色体上に散在し、これらの領域をインプリンティング領域といいます。インプリンティング領域内には、CpG配列に存在するシトシン(C)の親由来特異的メチル化が異なっているメチル化可変領域(DMR: differentially methylated region)が存在します(図1)。DMRのメチル化状態は、領域内のインプリンティング遺伝子の親由来特異的発現パターンと相関し、DMRのメチル化の異常は、インプリンティング遺伝子の発現異常を引き起こします。DMRは50か所報告されており、インプリンティング遺伝子の転写開始点周辺に存在することが多いのですが、インプリンティング遺伝子間やインプリンティング遺伝子のイントロンにも存在します(Monk et al. Epigenetics. 2018)。親由来のメチル化パターンが生殖細胞の段階で決定しているgermline DMRは、卵子でCpG配列がメチル化される34か所のDMRと精子でメチル化される3か所のDMRから構成されています。germline DMRは組織に関わらずDMRとなっていますが、germline DMRのメチル化状態に基づいて、受精後にメチル化パターンが決定するsecondary DMRは、組織によっては親由来で異なるメチル化パターンを示さず低メチル化となっている(胎盤など)こともあります。
Germline DMRのメチル化パターンは、図2に示すように受精後早期のゲノム全体の脱メチル化から守られています。DMRのメチル化の維持には、受精後ごく早期には主に卵子の中に存在しているタンパク質(NLRP2, NLRP5, NLRP7, PADI6など)が、引き続き胎児由来のタンパクであるZNF445やZFP57が重要な役割を果たしています(Eggermann et al. Nat rev prim dis. 2023)。
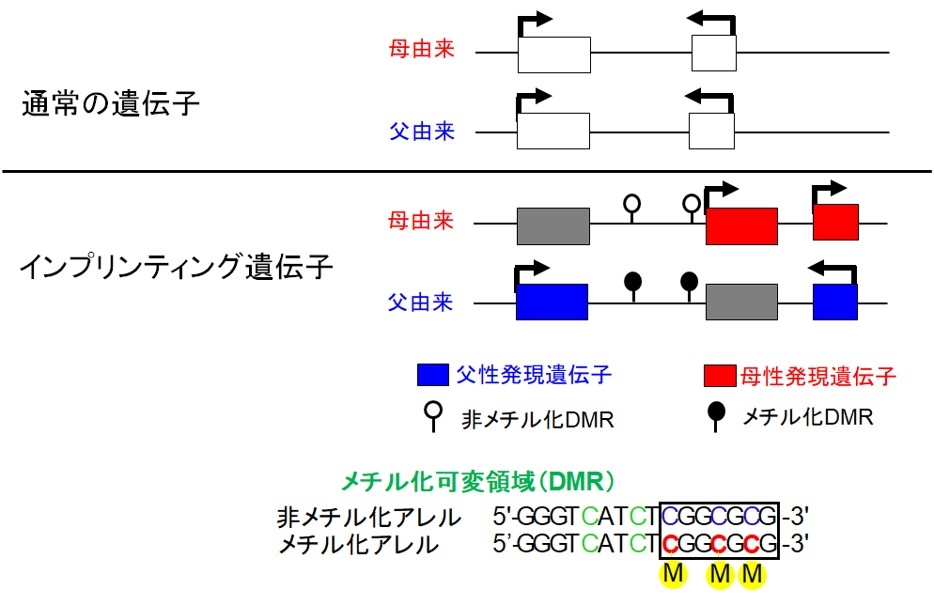
図1. インプリンティング領域
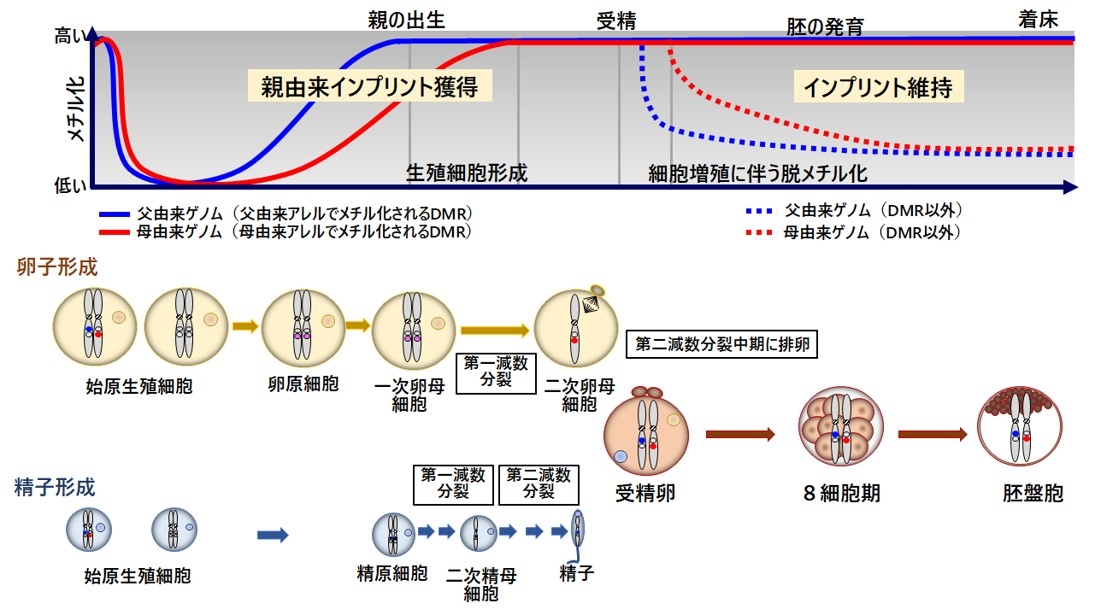
図2. 生殖細胞の成熟とインプリント獲得と維持
インプリンティング疾患の発症機序
インプリンティング疾患はインプリンティング遺伝子の発現異常を引き起こす遺伝学的原因(図3)により発症します。インプリンティング疾患の遺伝学的原因は、DMRを巻き込んだ異常と巻き込んでいない異常に大別され、前者には、片親性ダイソミー、エピ変異、DMRを含む欠失・重複が、後者にはDMRを含まない欠失・重複や遺伝子内変異が含まれます。
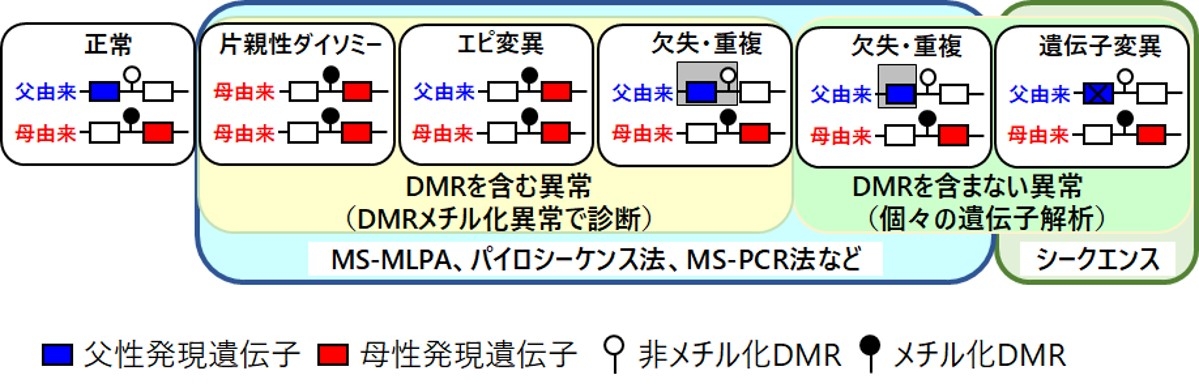
図3. インプリンティング疾患の遺伝学的原因
片親性ダイソミー (UPD: uniparental disomy)
相同染色体の全てあるいは一部分が、共に片親に由来する状態です。ともに母に由来する母性片親性ダイソミーを図3に示します。母性片親性ダイソミーでは、母性発現遺伝子は2コピー発現し、父性発現遺伝子の発現はなくなっており、インプリンティング遺伝子の発現異常が生じています(図3)。片親性ダイソミーには片親由来の一対の相同染色体からなるヘテロダイソミー (heterodisomy) と、相同染色体のうちの一本が伝わりそれが倍化したアイソダイソミー (isodisomy) があります(図4)。片親性ダイソミーの発症機序には大きく4つの機構があります。
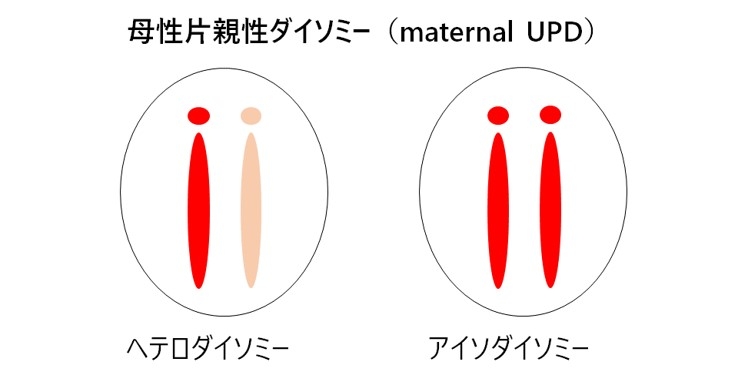
図4. ヘテロダイソミーとアイソダイソミー
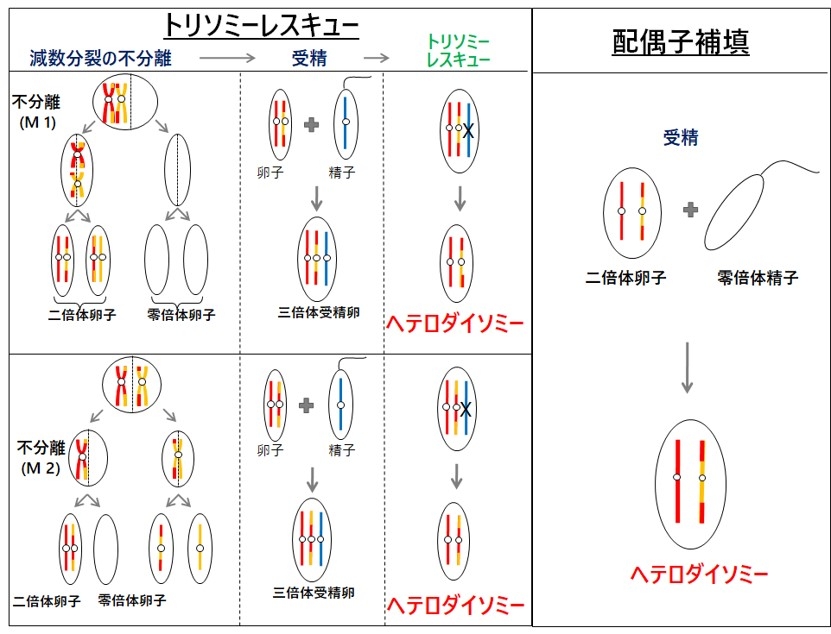
図5. トリソミーレスキューと配偶子補填
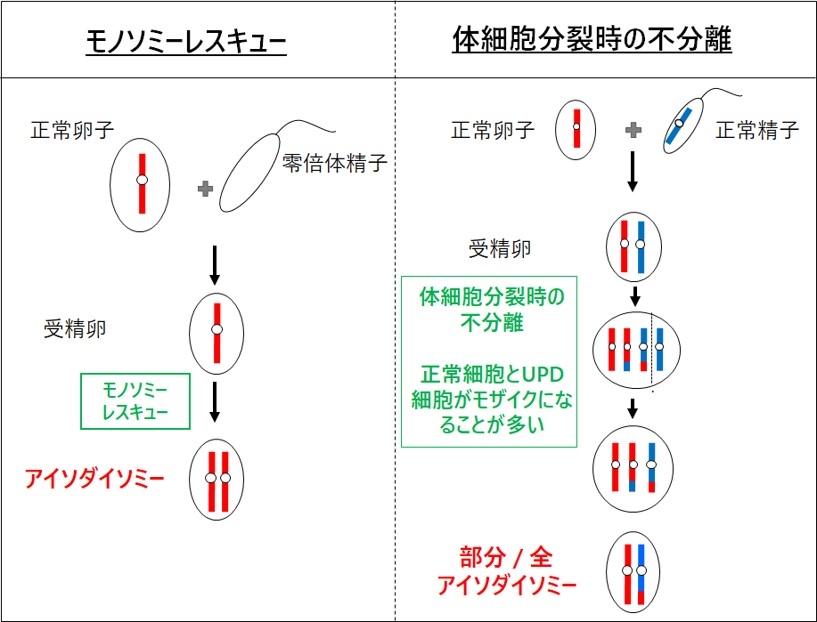
図6.モノソミーレスキューと体細胞分裂時の不分離
第1はトリソミーレスキュー(trisomy rescue)を介在した機構です(図5)。相同染色体が3本となるトリソミーは、21番染色体、13番染色体、18番染色体と性染色体を除いて出生することはできませんが、過剰染色体が脱落するトリソミーレスキューにより胎児の生存は可能となります。このとき、3分の1の確率で、片親性ダイソミーが発症します。トリソミーレスキューにより生じたUPDでは、相同染色体の組み換えによりヘテロダイソミーの領域とアイソダイソミーの領域が混在する。トリソミーは、染色体数の異常である配偶子(卵子や精子)が受精することにより生じます。21トリソミー(ダウン症)同様に、トリソミーレスキューを介したUPDにおいて母親の年齢が高いことが報告されています(Hara-Isono et al. Clinical Epigenet. 2023)。第2の発症機構は配偶子補填です。これは、ダイソミーとモノソミーの配偶子が受精することで生じますが、確率的に非常に低いと考えられます。第3の発症機構は、モノソミーレスキューです。染色体が一本しかないモノソミー細胞は、X染色体以外生存できませんが、モノソミーの染色体が複製されたときには生存可能になります。この場合、全ての染色体領域がアイソダイソミーとなります。第4の発症機構は、受精後の体細胞分裂時の不分離です。受精後の異常によるので、正常細胞とUPD細胞とのモザイクとなっていることが多く、しばしば部分的ダイソミーとなります。なお、片親性アイソダイソミーでは、片親が保因していた潜性変異の顕在化し疾患発症を招くことがあるので、遺伝性疾患の合併にも注意が必要です。
エピ変異
UPDやDMRを含む構造異常を認めずDMRのメチル化異常を示す場合、エピ変異と定義されます。DMR内のCpGは親由来依存性に片方のアレルではメチル化を受けず、もう一方のアレルではメチル化を受けることから、正常の場合、CpGのシトシンは50%程度のメチル化比率を示します。エピ変異の場合、メチル化を受けるはずのCpGのシトシンがメチル化を受けず異常低メチル化状態になる場合と、メチル化を受けないはずのCpGのシトシンがメチル化され異常高メチル化状態になる場合があります。エピ変異は遺伝子の発現異常を引き起こします。図3の場合、本来メチル化されない父由来染色体のDMRでメチル化されることにより母由来染色体と同様のメチル化パターンとなり、両アレルから母性発現遺伝子が発現し、父性発現遺伝子の発現が消失します。
さらに、最近の解析手法の進展により、多座位においてメチル化異常を呈するMLID (multilocus imprinting disturbance) という現象が、インプリンティング疾患患者の一部において同定されています。MLIDの原因は大部分で不明ですが、一部の患者さんでDMRのメチル化状態を保持するタンパク複合体を構成する分子の遺伝子(NLRP2, NLRP5, NLRP7, PADI6、ZFP57、ZNF445など)の変異が同定されています(Eggermann et al. Nat rev prim dis. 2023; Urakawa T et al. Clinical Epigenet. 2024)。
DMRを含むインプリンティング領域の欠失・重複
メチル化を受けないDMRは、領域内のインプリンティング遺伝子の発現制御領域 (IC: imprinting center) として機能しますが、メチル化を受けたDMRはICとして機能しません。したがって、非メチル化DMRを含む欠失は、そのアレル上のインプリンティング遺伝子の発現パターンを、メチル化DMRをもつアレルの遺伝子発現パターンに変え、結果的に遺伝子発現異常を引き起こします。一方、メチル化DMRを含む欠失は、もともとメチル化DMRはICとして機能していなかったことから、欠失領域内に発現しているインプリンティング遺伝子を含んでいない場合は、遺伝子発現異常を引き起こしません。
DMRを含まないインプリンティング領域の欠失・重複
欠失や重複が、DMRを含んでいなくてもインプリンティング疾患発症責任遺伝子を巻き込んでいるときに遺伝子発現異常が生じることからインプリンティング疾患が発症します。代表例は、プラダーウイリ症候群における疾患発症責任遺伝子SNORD116周辺のみの欠失です。
インプリンティング遺伝子内変異
疾患責任インプリンティング遺伝子に変異がある場合、インプリンティング疾患が発症します。例えば、ベックウィズ-ヴィーデマン症候群の責任遺伝子である母性発現遺伝子CDKN1Cにおいて、母由来アレル上のCDKN1Cに変異がある場合、ベックウィズ-ヴィーデマン症候群が発症しますが、父由来アレル上のCDKN1Cに変異がある場合は発症しません。
主要なインプリンティング疾患
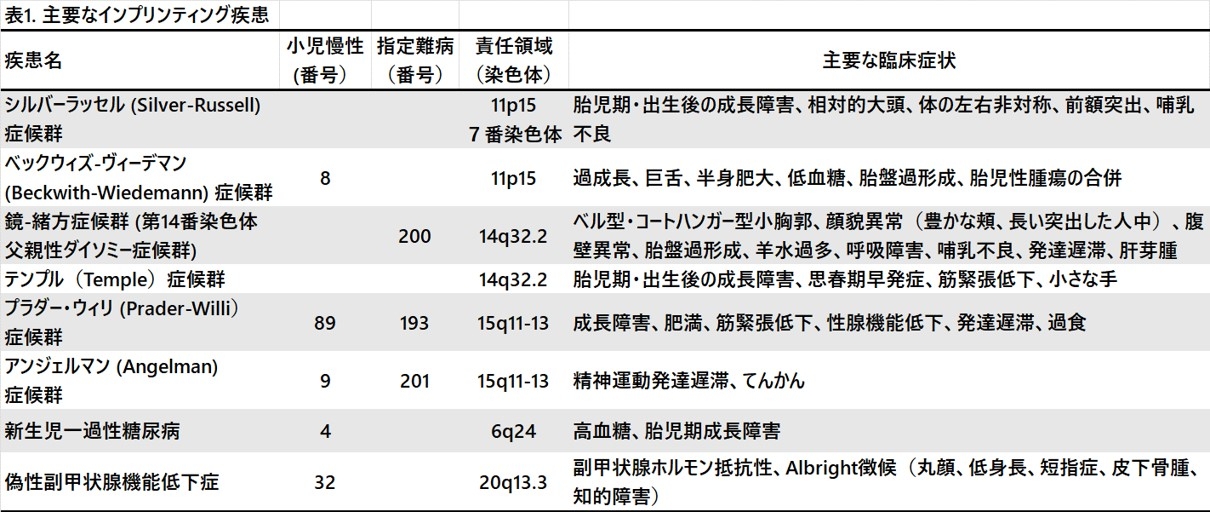
疾患概念が確立した主要なインプリンティング疾患は8疾患あります(表1)。インプリンティング遺伝子は成長に関係する機能をもっていることが多く、インプリンティング疾患では成長障害や過成長といった成長の異常を認めることが多いです。また、疾患間での症状のオーバーラップも認められています。
遺伝子診断方法
図3に示したように、DMRのメチル化テストでスクリーニングできる場合と、メチル化異常を認めず変異解析を必要とする場合があります。保険収載されている遺伝子診断法を表2に記します。プラダー・ウィリ症候群、アンジェルマン症候群、鏡-緒方症候群を診断するための遺伝子診断法には、DMRのメチル化状態を調べるメチル化テストおよびメチル化状態とコピー数異常を検出できるMS-MLPA (Multiplex Ligation-dependent Probe Amplification) 法があり、これらは保険収載されています。現時点では国立成育医療研究センター衛生検査センターのみがMS-MLPA法による臨床検査を受託しています。なお、この検査を保険収載された臨床検査として実施するには、依頼元の医療施設が施設基準を満たす必要があります。施設基準については、厚生労働省などから公表される診療報酬点数表や関連する通知https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iryouhoken/newpage_21053.html などをご参照ください。
- コピー数異常とホモ接合性領域ROH (Region of Homozygosity) を検出できるマイクロアレイ染色体検査(染色体構造変異解析)は、プラダー・ウィリ症候群、アンジェルマン症候群、鏡-緒方症候群に加え、ベックウィズ-ヴィーデマン症候群、シルバー・ラッセル症候群、テンプル症候群で保険収載されています。図3に示す通り、インプリンティング疾患はDMRのメチル化異常をきたすことが大部分であることから、スクリーニング検査としては、メチル化の異常とコピー数の異常を同定できるMS-MLPA法が推奨されています(Mackay et al. Clinical Epigenetics. 2022)。マイクロアレイ染色体検査は全染色体のコピー数異常を検出できますが、インプリンティング領域内の数10 kbより小さい構造異常は検出できません。また、この検査では、アイソダイソミーによるUPDを検出することができますが、ヘテロダイソミーによるUPDは検出できません。よって、保険収載されていますが、インプリンティング疾患を疑うとき、マイクロアレイ染色体検査は最初に行うべき検査ではありません。
